Licence CC BY-NC-ND - Thierry Parmentelat
visu: DNA Walking¶
Nous allons voir dans ce complément une version exécutable de l’algorithme dit de DNA Walking pour représenter un brin de DNA en 2 dimensions.
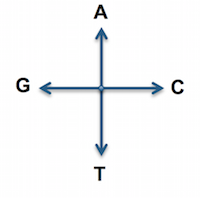
Il s’agit donc de dessiner le parcours d’une séquence d’ADN, en décidant que chaque nucléotide C, A, G, et T correspond à une direction dans le plan:
La librairie matplotlib¶
La librairie que nous allons utiliser pour dessiner le chemin s’appelle matplotlib, principalement parce qu’elle est d’un usage très répandu pour mettre en forme des résultats de calcul.
# pour des graphiques interactifs - version notebook seulement
# on peut envisager de faire
#%matplotlib ipympl
# mais pour la sortie HTML les affichages sont statiques
# importation de la librairie
import matplotlib.pyplot as plt
# enfin: la taille à utiliser pour les figures
import pylab
pylab.rcParams['figure.figsize'] = (6, 6)matplotlib va pouvoir très facilement dessiner le chemin qui nous intéresse, si on lui fournit deux listes de valeurs, qu’on appelle en général simplement X et Y, de même taille, et qui vont contenir les coordonnées des points par lesquels on passe.
Voyons cela tout de suite sur un exemple construit “à la main”: imaginons que l’on veuille dessiner un chemin qui passe par :
premier point (0, 0)
deuxième point (2, 1)
troisième point (1, 0)
quatrième point (3, 4)
# on construit la liste des abscisses
X = [0, 2, 1, 3]
# et la liste des ordonnées
Y = [0, 1, 0, 4]Et pour dessiner il nous suffit alors d’appeler la fonction plot comme ceci :
plt.figure()
plt.plot(X, Y, 'r')
plt.show()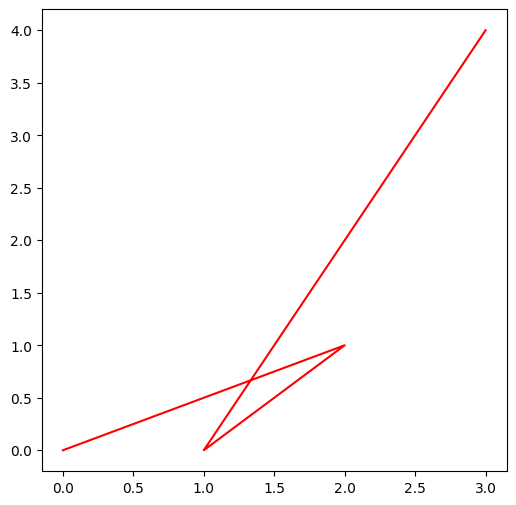
Des fonctions qui renvoient deux valeurs¶
Donc pour dessiner un fragment d’ADN, le problème revient simplement à calculer les coordonnées des points du chemin, sous la forme d’une liste d’abscisses et une liste d’ordonnées.
Nous sommes donc confrontés au besoin d’écrire une fonction, mais qui doit renvoyer deux choses (la liste des abscisses et la liste des ordonnées), et idéalement en une seule passe pour être aussi efficace que possible.
Il est très facile en python de renvoyer plusieurs valeurs dans une fonction. Voyons ça sur un premier exemple très simple : une fonction qui calcule le carré et le cube d’un nombre.
# une fonction qui renvoie deux valeurs
def square_and_cube(x):
carre = x * x
cube = x ** 3
# techniquement : on construit un tuple avec ces deux valeurs
return carre, cubePour utiliser les deux résultats de la fonction, on utilise tout simplement cette syntaxe :
a, b = square_and_cube(5)
print("a =", a)
print("b =", b)a = 25
b = 125
Utiliser un dictionnaire¶
Avant de voir le parcours de l’ADN à proprement parler, il nous reste à décider comment représenter l’association
entre d’une part les 4 lettres de notre alphabet
C,A,GetT,et de l’autre les déplacements correspondants dans le plan.
Pour cela, il est naturel en Python d’utiliser un dictionnaire, qui d’associer des valeurs à des clés comme ceci :
moves = {
'C' : [1, 0],
'A' : [0, 1],
'G' : [-1, 0],
'T' : [0, -1],
}De sorte que par exemple on pourra facilement calculer le déplacement à faire lorsqu’on voit passer un C :
moves['C'][1, 0]Ce qui signifie pour nous que lorsqu’on rencontre un C, il faut :
faire
+1en x,et ne rien faire (ajouter
0) en y.
Que l’on peut écrire, en utilisant la même syntaxe que tout à l’heure :
delta_x, delta_y = moves['C']
print("à ajouter en x: ", delta_x)
print("à ajouter en y: ", delta_y)à ajouter en x: 1
à ajouter en y: 0
Le parcours à proprement parler¶
Nous avons à présent tous les éléments pour écrire une fonction, qui
prend en entrée un fragment d’ADN codé comme une chaine de caractères contenant les 4 abbréviations,
et qui retourne deux listes, correspondant aux X et aux Y respectivement, des points du chemin.
# un algorithme qui calcule les deux chemins en x et y
# en partant et en se deplaçant le long de la chaine
def path_x_y(adn):
# initialise les résultats
path_x, path_y = [], []
# on commence au centre
x, y = 0, 0
# le point de départ fait partie du chemin
path_x.append(x)
path_y.append(y)
# pour tout l'ADN
for nucleotide in adn:
# quel deplacement faut-il faire
delta_x, delta_y = moves[nucleotide]
# on l'applique
x += delta_x
y += delta_y
# on range le point courant
# dans les listes resultat
path_x.append(x)
path_y.append(y)
return path_x, path_yVoyons ce que cela nous donne sur un tout petit brin d’ADN pour commencer :
small_adn = "CAGACCACT"
X, Y = path_x_y(small_adn)
print("les abscisses", X)les abscisses [0, 1, 1, 0, 0, 1, 2, 2, 3, 3]
plt.figure()
plt.plot(X, Y, 'r')
plt.show()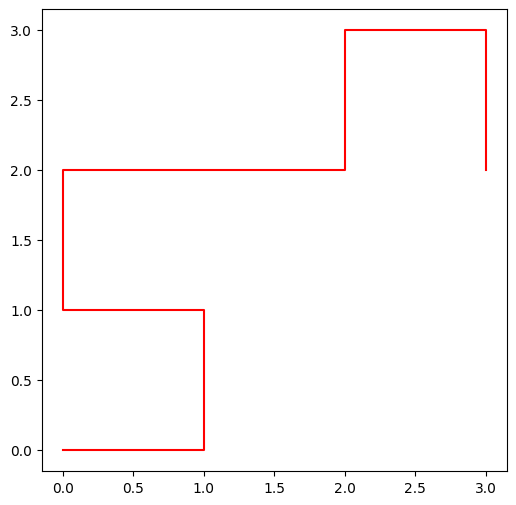
Un raccourci¶
Si on veut tout mettre ensemble dans une seule fonction plus pratique à appeler :
def walk(adn):
plt.figure()
print("longueur de la séquence d'entrée", len(adn))
X, Y = path_x_y(adn)
plt.plot(X, Y, 'r')
plt.show()walk(small_adn)longueur de la séquence d'entrée 9
Des données plus grosses¶
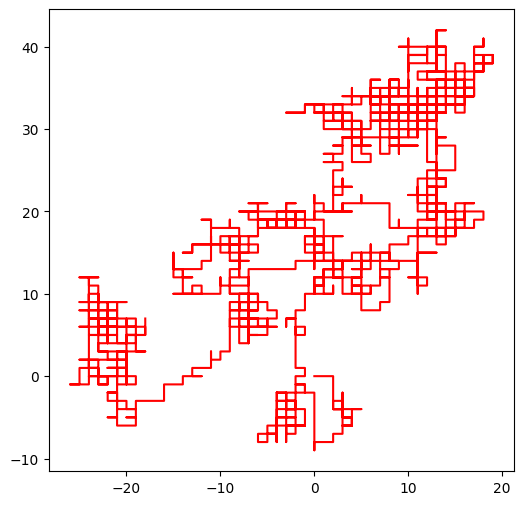
Si on prend par exemple le brin d’ADN qui est illustré dans le transparent de la séquence 7 :
from adn_samples import sample_week1_sequence7
print(sample_week1_sequence7)CCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGAACTGGTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATACTTTAACCAATATAGGCATAGCGCACAGACAGATAAAAATTACAGAGTACACAACATCCATGAAACGCATTAGCACCACCATTACCACCACCATCACCATTACCACAGGTAACGGTGCGGGCTGACGCGTACAGGAAACACAGAAAAAAGCCCGCACCTGACAGTGCGGGCTTTTTTTTTCGACCAAAGGTAACGAGGTAACAACCATGCGAGTGTTGAAGTTCGGCGGTACATCAGTGGCAAATGCAGAACGTTTTCTGCGTGTTGCCGATATTCTGGAAAGCAATGCCAGGCAGGGGCAGGTGGCCACCGTCCTCTCTGCCCCCGCCAAAATCACCAACCACCTGGTGGCGATGATTGAAAAAACCATTAGCGGCCAGGATGCTTTACCCAATATCAGCGATGCCGAACGTATTTTTGCCGAACTTTTGACGGGACTCGCCGCCGCCCAGCCGGGGTTCCCGCTGGCGCAATTGAAAACTTTCGTCGATCAGGAATTTGCCCAAATAAAACATGTCCTGCATGGCATTAGTTTGTTGGGGCAGTGCCCGGATAGCATCAACGCTGCGCTGATTTGCCGTGGCGAGAAAATGTCGATCGCCATTATGGCCGGCGTATTAGAAGCGCGCGGTCACAACGTTACTGTTATCGATCCGGTCGAAAAACTGCTGGCAGTGGGGCATTACCTCGAATCTACCGTCGATATTGCTGAGTCCACCCGCCGTATTGCGGCAAGCCGCATTCCGGCTGATCACATGGTGCTGATGGCAGGTTTCACCGCCGGTAATGAAAAAGGCGAACTGGTGGTGCTTGGACGCAACGGTTCCGACTACTCTGCTGCGGTGCTGGCTGCCTGTTTACGCGCCGATTGTTGCGAGATTTGGACGGACGTTGACGGGGTCTATACCTGCGACCCGCGTCAGGTGCCCGATGCGAGGTTGTTGAAGTCGATGTCCTACCAGGAAGCGATGGAGCTTTCCTACTTCGGCGCTAAAGTTCTTCACCCCCGCACCATTACCCCCATCGCCCAGTTCCAGATCCCTTGCCTGATTAAAAATACCGGAAATCCTCAAGCACCAGGTACGCTCATTGGTGCCAGCCGTGATGAAGACGAATTACCGGTCAAGGGCATTTCCAATCTGAATAACATGGCAATGTTCAGCGTTTCTGGTCCGGGGATGAAAGGGATGGTCGGCATGGCGGCGCGCGTCTTTGCAGCGATGTCACGCGCCCGTATTTCCGTGGTGCTGATTACGCAATCATCTTCCGAATACAGCATCAGTTTCTGCGTTCCACAAAGCGACTGTGTGCGAGCTGAACGGGCAATGCAGGAAGAGTTCTACCTGGAACTGAAAGAAGGCTTACTGGAGCCGCTGGCAGTGACGGAACGGCTGGCCATTATCTCGGTGGTAGGTGATGGTATGCGCACCTTGCGTGGGATCTCGGCGAAATTCTTTGCCGCACTGGCCCGCGCCAATATCAACATTGTCGCCATTGCTCAGGGATCTTCTGAACGCTCAATCTCTGTCGTGGTAAATAACGATGATGCGACCACTGGCGTGCGCGTTACTCATCAGATGCTGTTCAATACCGATCAGGTTATCGAAGTGTTTGTGATTGGCGTCGGTGGCGTTGGCGGTGCGCTGCTGGAGCAACTGAAGCGTCAGCAAAGCTGGCTGAAGAATAAACATATCGACTTACGTGTCTGCGGTGTTGCCAACTCGAAGGCTCTGCTCACCAATGTACATGGCCTTAATCTGGAAAACTGGCAGGAAGAACTGGCGCAAGCCAAAGAGCCGTTTAATCTCGGGCGCTTAATTCGCCTCGTGAAAGAATATCATCTGCTGAACCCGGTCATTGTTGACTGCACTTCCAGCCAGGCAGTGGCGGATCAATATGCCGACTTCCTGCGCGAAGGTTTCCACGTTGTCACGCCGAACAA
On peut le dessiner tout simplement comme ceci :
walk(sample_week1_sequence7)longueur de la séquence d'entrée 2071
Le résultat sur de vraies séquences¶
Si vous allez vous promener sur http://
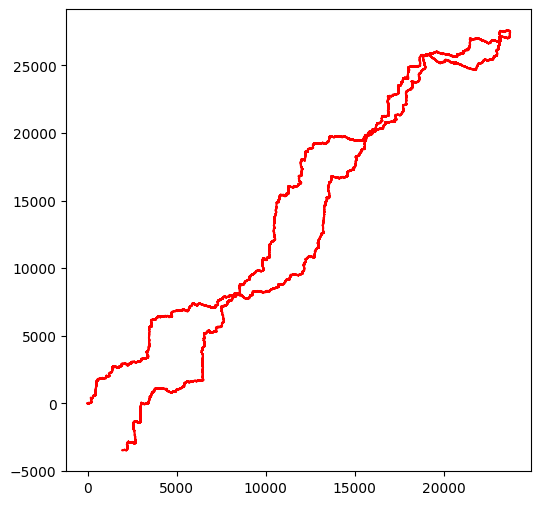
Un point de rebroussement très visible : Borrelia¶
Pour le premier exemple nous allons regarder le résultat de notre visualisation avec Borrelia, que vous pouvez consulter ici, ou retrouver en entrant dans http://CP000013. Nous l’avons chargé pour vous (voir plus loin comment vous pouvez charger d’autres spécimens à partir d’une autre clé) :
from adn_samples import borrelia
print("taille de borrelia", len(borrelia))taille de borrelia 904246
Avec cet échantillon vous allez voir très distinctement un point de rebroussement apparaître :
walk(borrelia)longueur de la séquence d'entrée 904246
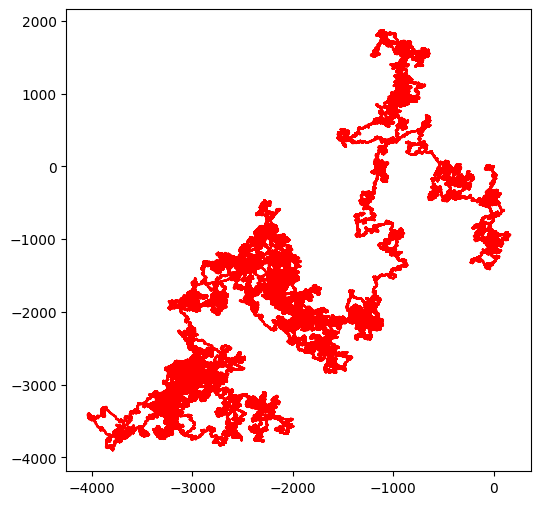
Un contrexemple : Synechosystis¶
A contrario, voici ce qu’on obtient avec Synechosystis (clé BA000022). Soyez patient car ce brin contient environ 3.5 millions de bases.
from adn_samples import synechosystis
walk(synechosystis)longueur de la séquence d'entrée 3573470
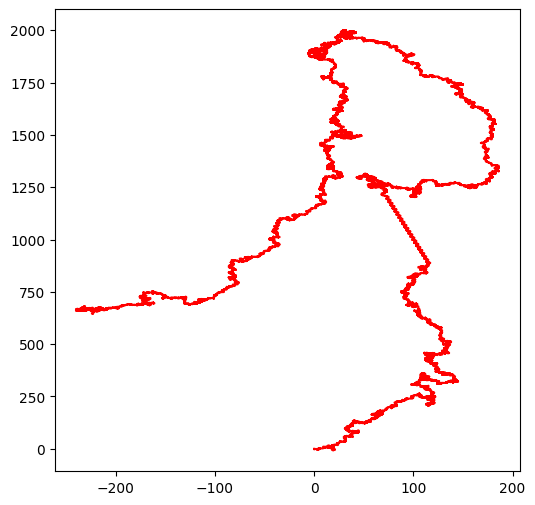
Des données réelles¶
Pour illustrer ce qu’il est possible de faire très simplement aujourd’hui, je suis allé sur le site du Eureopan Nucleotide Archive j’ai cherché “Borrelia burgdorferi B31” et je suis arrivé à cette page :
http://
Nous vous fournissons un petit utilitaire (très sommaire) qui permet d’aller chercher de telles séquences pour les manipuler directement dans ce notebook :
import adn_fetchQue vous pouvez utiliser comme ceci à partir de la clé de la séquence qui vous intéresse :
burgdorferi = adn_fetch.fetch('AE000789')
burgdorferidownload http://www.ebi.ac.uk/ena/browser/api/embl/AE000789 returns 60120 chars
# et que maintenant on peut donc dessiner aussi
walk(burgdorferi)longueur de la séquence d'entrée 27323
Explorer le chemin de manière interactive¶
(version notebook seulement: si vous avez activé %matplotlib ipympl): remarquez sur la vue les boutons pour la navigation interactive
la maison: pour retourner à la vue initiale
le carré: pour zoomer
...